
Reliable Drug–Target Interaction Prediction Using Convolutional Neural Networks with Robust Negative Sample Generation
Keywords:
Drug–target interaction (DTI) prediction, Convolutional neural networks (CNNs), Area under the ROC curve (AUC), Negative instancesAbstract
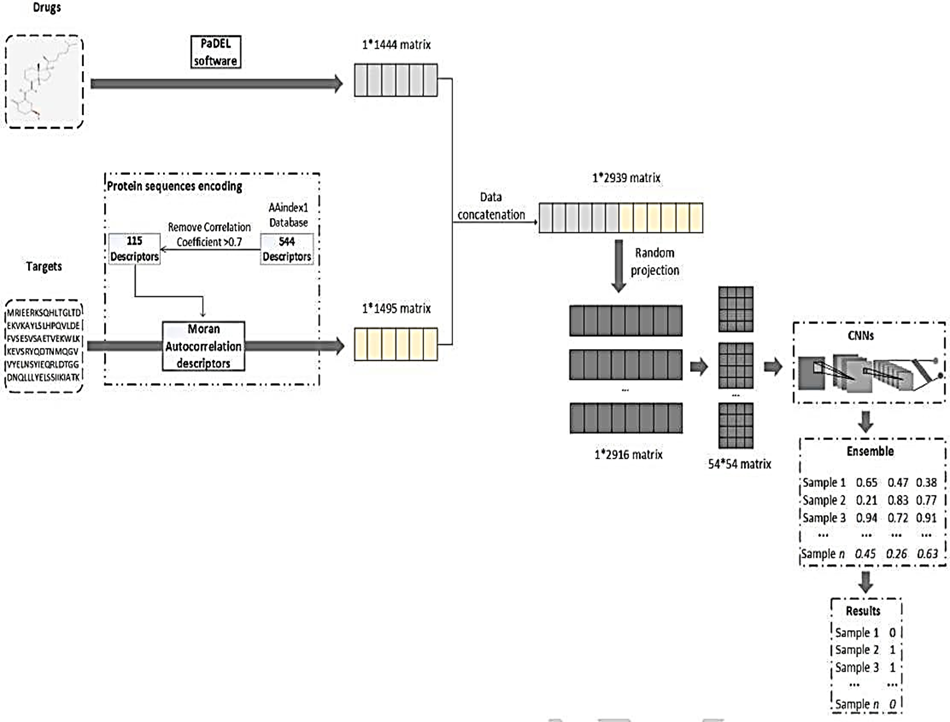
- Proteins, including receptors, enzymes, and ion channels, represent primary biological targets whose interactions with small-molecule drugs play a critical role in therapeutic discovery and development. Accurate identification of drug–target interactions (DTIs) remains a fundamental challenge in drug discovery due to the high cost, time requirements, and scalability limitations of experimental validation. Consequently, computational approaches have emerged as efficient alternatives for large-scale DTI prediction. This study proposes a convolutional neural network (CNN) based framework for predicting drug–target interactions, with a particular focus on reliable negative sample generation to address the inherent data imbalance and uncertainty present in DTI datasets. The proposed method incorporates feature projection techniques to effectively capture meaningful representations of drug and protein features while reducing noise and redundancy. By constructing more reliable negative instances, the framework improves model robustness and mitigates bias commonly introduced by randomly generated negative samples. The proposed model is evaluated on a benchmark DTI dataset, where it achieves a classification accuracy of 0.9800, demonstrating strong predictive capability. To further assess generalization performance, the model is tested on an independent external dataset derived from DrugBank. On this dataset, the framework attains an accuracy of 0.8814 and an Area Under the Receiver Operating Characteristic Curve (AUC) of 0.9527, indicating effective transferability across datasets. Experimental results confirm that the integration of CNN-based feature learning with reliable negative instance generation significantly enhances DTI prediction performance. The proposed framework offers a robust and generalizable computational tool for drug–target interaction prediction and has the potential to support early-stage drug discovery by reducing experimental search space and accelerating candidate prioritization.
Downloads
References
[1] L. Wang, Z. H. You, X. Chen, X. Yan, G. Liu, and W. Zhang, “Rfdt: A rotation forest-based predictor for predicting drug-target interactions using drug structure and protein sequence information,” Current Protein Peptide Science, vol. 19, pp. 445–454, 2016.
[2] B. Booth and R. Zemmel, “Prospects for productivity,” Nature Reviews Drug Discovery, vol. 3, no. 5, pp. 451–456, 2004.
[3] Y.-A. Huang, Z.-H. You, and X. Chen, “A systematic prediction of drug-target interactions using molecular fingerprints and protein sequences,” Current Protein & Peptide Science, vol. 19, pp. 468–478, 2018.
[4] A. L. Hopkins, “Network pharmacology: The next paradigm in drug discovery,” Nature Chemical Biology, vol. 4, no. 11, pp. 682–690, 2008.
[5] M. J. Keiser, B. L. Roth, B. N. Armbruster, P. Ernsberger, J. J. Irwin, and B. K. Shoichet, “Relating protein pharmacology by ligand chemistry,” Nature Biotechnology, vol. 25, no. 2, pp. 197–206, 2007.
[6] R. Iorio, R. Shrestha, M. Berube, and A. R. Licinio, “Pathway analysis of polypharmacology,” Briefings in Bioinformatics, vol. 15, no. 2, pp. 278–289, 2014.
[7] D. Lounkine, M. J. Keiser, S. Whitebread, D. Mikhailov, J. Hamon, J. L. Jenkins, P. Lavan, E. Weber, A. K. Doak, S. Côté, B. K. Shoichet, and L. Urban, “Large-scale prediction and testing of drug activity on side-effect targets,” Nature, vol. 486, pp. 361–367, 2012.
[8] M. Campillos, M. Kuhn, A.-C. Gavin, L. J. Jensen, and P. Bork, “Drug target identification using side-effect similarity,” Science, vol. 321, no. 5886, pp. 263–266, 2008.
[9] X. Chen, H. Y. Ji, G. Y. Yan, and L. Y. Han, “Drug–target interaction prediction: databases, web servers and computational models,” Briefings in Bioinformatics, vol. 17, no. 4, pp. 696–712, 2016.
[10] Z. Li, P. Han, and J. Lin, “A machine learning based method for predicting drug–target interactions using drug fingerprints and protein sequence descriptors,” Molecular BioSystems, vol. 12, no. 7, pp. 2431–2439, 2016.
[11] B. R. Donald, Algorithms in Structural Molecular Biology, Cambridge, MA, USA: MIT Press, 2011.
[12] G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell, and A. J. Olson, “AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility,” Journal of Computational Chemistry, vol. 30, no. 16, pp. 2785–1795, 2009.
[13] A. C. Cheng, R. G. Coleman, K. T. Smyth, Q. Cao, P. Soulard, D. R. Caffrey, A. C. Salzberg, and E. S. Huang, “Structure-based maximal affinity model predicts small-molecule druggability,” Nature Biotechnology, vol. 25, no. 1, pp. 71–75, 2007.
[14] Y. Yamanishi, M. A. Araki, W. Honda, and M. Kanehisa, “Prediction of drug-target interaction networks from the integration of chemical and genomic spaces,” Bioinformatics, vol. 24, no. 13, pp. i232–i240, 2008.
[15] Y.-Y. Wang, J. C. Nacher, and X.-M. Zhao, “Predicting drug targets based on protein domains,” Molecular Biosystems, vol. 8, pp. 1528–1534, Apr. 2012.
[16] X. Chen, M. X. Liu, and G. Y. Yan, “Drug-target interaction prediction by random walk on the heterogeneous network,” Molecular BioSystems, vol. 8, no. 7, pp. 1970–1978, 2012.
[17] H. Yu, J. Chen, X. Xu, Y. Li, H. Zhao, Y. Fang, X. Li, W. Zhou, W. Wang, and Y. Wang, “A systematic prediction of multiple drug-target interactions from chemical, genomic, and pharmacological data,” PLoS ONE, vol. 7, no. 5, 2012.
[18] Y. Wang and J. Zeng, “Predicting drug-target interactions using restricted Boltzmann machines,” Bioinformatics, vol. 29, no. 13, pp. 126–134, 2013.
[19] F.-R. Meng, Z.-H. You, X. Chen, Y. Zhou, and J.-Y. An, “Prediction of drug-target interaction networks from the integration of protein sequences and drug chemical structures,” Molecules, vol. 22, no. pii: E1119, Jul. 2017.
[20] W. Ming, Z. Zhang, S. Niu, H. Sha, R. Yang, Y. Yun, and H. Lu, “Deep-learning-based drug-target interaction prediction,” Journal of Proteome Research, vol. 16, no. 4, 2017.
[21] M. Hamanaka, K. Taneishi, H. Iwata, J. Ye, J. Pei, J. Hou, and Y. Okuno, “Cgbvsdnn: Prediction of compound-protein interactions based on deep learning,” Molecular Informatics, vol. 36, no. 1/2, 2017.
[22] F. Wan and J. Zeng, “Deep learning with feature embedding for compound-protein interaction prediction,” 2016.
[23] G. E. Hinton and R. R. Salakhutdinov, “Reducing the dimensionality of data with neural networks,” Science, vol. 313, pp. 504–507, Jul. 2006.
[24] G. E. Hinton, S. Osindero, and Y.-W. Teh, “A fast learning algorithm for deep belief nets,” Neural Computation, vol. 18, no. 7, pp. 1527–1554, 2006.
[27] M. Kanehisa, Y. Sato, M. Kawashima, M. Furumichi, and M. Tanabe, “KEGG as a reference resource for gene and protein annotation,” Nucleic Acids Research, vol. 44, no. D1, pp. D457–D462, 2016.
[28] D. S. Wishart, Y. D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, T. Sajed, D. Johnson, C. Li, Z. Sayeeda, and A. Assempour, “DrugBank 5.0: a major update to the DrugBank database for 2018,” Nucleic Acids Research, vol. 46, no. D1, pp. D1074–D1082, 2018.
[29] Z. He, J. Zhang, X. H. Shi, L. L. Hu, X. Kong, Y. D. Cai, and K. C. Chou, “Predicting drug-target interaction networks based on functional groups and biological features,” PLoS ONE, vol. 5, no. 3, 2010.
[30] T. U. Consortium, “UniProt: The universal protein knowledgebase,” Nucleic Acids Research, vol. 45, no. D1, pp. D158–D169, 2017.
[31] N. Shaikh, M. Sharma, and P. Garg, “An improved approach for predicting drug-target interaction: Proteochemometrics to molecular docking,” Molecular BioSystems, vol. 12, no. 3, 2016.
[32] C. W. Yap, “Padel-descriptor: An open-source software to calculate molecular descriptors and fingerprints,” Journal of Computational Chemistry, vol. 32, no. 7, pp. 1466–1474, 2011.
[33] S. Kawashima and M. Kanehisa, “Aaindex: Amino acid index database,” Nucleic Acids Research, vol. 27, no. 1, pp. 368–369, 1999.
[34] Z. R. Li, H. H. Lin, L. Y. Han, L. Jiang, X. Chen, and Y. Z. Chen, “Profeat: A web server for computing structural and physico-chemical features of proteins and peptides from amino acid sequence,” Nucleic Acids Research, vol. 39, no. Web Server issue, 2011.
[35] Z. R. Li, L. Y. Han, L. Jiang, X. Chen, and Y. Z. Chen, “Prediction of subcellular location of mycobacterial proteins using feature fusion and support vector machine,” Journal of Proteome Research, vol. 5, no. 11, pp. 2780–2788, 2006.
[36] A. Krizhevsky, I. Sutskever, and G. E. Hinton, “ImageNet classification with deep convolutional neural networks,” Communications of the ACM, vol. 60, no. 6, pp. 84–90, 2017.
[37] K. Simonyan and A. Zisserman, “Very deep convolutional networks for large-scale image recognition,” International Conference on Learning Representations (ICLR), 2015.
[38] Y. LeCun, L. Bottou, Y. Bengio, and P. Haffner, “Gradient-based learning applied to document recognition,” Proceedings of the IEEE, vol. 86, no. 11, pp. 2278–2324, 1998.
[39] K. He, X. Zhang, S. Ren, and J. Sun, “Delving deep into rectifiers: Surpassing human-level performance on ImageNet classification,” Proceedings of the IEEE International Conference on Computer Vision (ICCV), pp. 1026–1034, 2015.
[40] M. D. Zeiler and R. Fergus, “Visualizing and understanding convolutional networks,” European Conference on Computer Vision (ECCV), Springer, pp. 818–833, 2014.
[41] Y. LeCun, B. Boser, J. S. Denker, D. Henderson, R. E. Howard, W. Hubbard, and L. D. Jackel, “Handwritten digit recognition with a back-propagation network,” Advances in Neural Information Processing Systems (NeurIPS), vol. 2, pp. 396–404, 1990.
[42] V. Nair and G. E. Hinton, “Rectified linear units improve restricted Boltzmann machines,” Proceedings of the 27th International Conference on Machine Learning (ICML), pp. 807–814, 2010.
[43] D. P. Kingma and J. Ba, “Adam: A method for stochastic optimization,” International Conference on Learning Representations (ICLR), 2015.
[44] N. Srivastava, G. Hinton, A. Krizhevsky, I. Sutskever, and R. Salakhutdinov, “Dropout: A simple way to prevent neural networks from overfitting,” Journal of Machine Learning Research, vol. 15, pp. 1929–1958, 2014.
[45] S. Ioffe and C. Szegedy, “Batch normalization: Accelerating deep network training by reducing internal covariate shift,” Proceedings of the 32nd International Conference on Machine Learning (ICML), pp. 448–456, 2015.
[46] S. Dasgupta and A. Gupta, “An elementary proof of the Johnson–Lindenstrauss lemma,” International Computer Science Institute Technical Report, 1999.
[47] Ahmed, Abdelmoty M., et al. "Enhancing Drug-Target Interaction Prediction with CNN-Based Deep Learning and Systematic Encoding Strategies." Journal of Smart Algorithms and Applications (JSAA) 1.1 (2025): 5-16.
Downloads
Published
Issue
Section
Categories
License
Copyright (c) 2026 Journal of Smart Algorithms and Applications (JSAA)

This work is licensed under a Creative Commons Attribution 4.0 International License.
Journal of Smart Algorithms and Applications (JSAA) content is published under a Creative Commons Attribution License (CCBY). This means that content is freely available to all readers upon publication, and content is published as soon as production is complete.
Journal of Smart Algorithms and Applications (JSAA) seeks to publish the most influential papers that will significantly advance scientific understanding. Selected articles must present new and widely significant data, syntheses, or concepts. They should merit recognition by the wider scientific community and the general public through publication in a reputable scientific journal.










